Highlights from the EMA’s finalised guidelines on computerised systems and electronic data in clinical trials’ – Part 1
On the 9th of March, the EMA published their finalised guidelines on computerised systems and electronic data in clinical trials. These give guidance on the use of computerised systems and electronic data to ensure data quality, reliability and integrity.
Regulations can be difficult to interpret, only instructing on the expected outcomes without guidance around how to get there. This approach can often cause confusion around how best to comply, risking non-compliance and ultimately hefty fines and delays.
The purpose of guidelines such as these, is to offer clearer advice to sponsors on specific regulations to better prepare for inspections and demonstrate compliance. This particular piece of guidance covers multiple regulations surrounding clinical trials such as EU Regulation 536/2014 (which will be referenced throughout this post).
For example, this particular regulation brought in the requirement to retain, and most importantly, ensure the long-term access and use of the trial master file (TMF) for +25 years. That said, there were limited details provided about exactly what is expected by sponsors when archiving these files for that length of time. If you’re interested in finding about more about these regulations specifically, you can find out more here.
This two-part blog series will focus on highlighting key elements from this set of guidelines. Part 1 will provide more detail on what inspectors are expecting when it comes to the digital archival of clinical trial records.
What do these guidelines refer to?
First of all, it’s crucial to understand what data these guidelines are referring to. This set of guidelines refer to all electronic data that is generated during clinical trials, whether this may be raw or processed data. This includes data captured from electronic Case Report Forms (eCRFs), electronic source data, electronic data capture (EDC) systems and other computer systems used. As well as this, it covers metadata associated with the electronic data, such as system logs, audit trails and other system-generated data that records activity.
Key retention elements within the guidelines:
1- ALCOA++ standard practice
Before we dive into the guidelines specific to retention requirements, a critical point to note within the document is that the ALCOA++ principles are highlighted as an expected practice in achieving data integrity. Specifically, it states that “data integrity is achieved when data are collected, accessed, and maintained in a secure manner, to fulfil the ALCOA++ principles.”
In short, this means that to maintain data integrity, the ALCOA++ principles must be followed for the entire lifecycle of clinical trial data, including during the +25 year retention period.
We recently published an eBook as a guide to achieving long-term data integrity using the ALCOA++ principles alongside maturity models (access it here).
2- “Suitable archiving systems should be in place to safeguard data integrity for the periods established by the regulatory requirements including those in any of the regions where the data may be used for regulatory submissions, and not just those of the country where the data are generated.”
The guidelines are very clear here; organisations must have a suitable archive system in place which can safeguard the integrity of the digital records for the entire retention period. This means ensuring that where you chose to archive your data, must be an archive and additionally must align to the ALCOA++ principles (as referenced in section 4.1 of the guidance documents and above).
To take the example of Legible from ALCOA, you must ensure that your records will be readable for the entirety of the retention period. For digital records, this means at a minimum ensuring that you are maintaining those records in long-term formats which can be opened and read by future devices. If you’re interested in reading more about what long-term data integrity in line with ALCOA++ looks like, the aforementioned eBook on the very topic provides more detail.
The other element to highlight from this point, is that this requirement applies to where the regulatory submission is being made, and not just where the data was generated. So, for example, if a trial was run in the United States, but the submission was made in Europe, then the sponsor would have to align with these requirements, and not just those outlined by the FDA.
3- “It should be ensured that the file and any software required (depending on the media used for storage) remain accessible, throughout the retention period. This could imply e.g. migration of data (see section 6.9.)”
Ensuring accessibility of data for long-term retention periods means that data needs to be safeguarded against risk of corruption, loss and obsolescence. Additionally, for records to be accessible they also need to be findable.
When maintaining access to long-term records, organisations need to consider things such as:
- Are records being maintained in long-term formats so they can be opened and read by future devices and software?
- Are you regularly checking each record for any loss or corruption?
- How are you managing access to those records over the 25+ years? Will you always have access to the current systems? And how would you change access in future if required?
- Are you attributing the right metadata to each record so that they can be found in future?
The guidance also references that this could imply that you might need to migrate your data to a difference system in future to retain access. This could be from a locked system to a dedicated archive, or moving data from one archive to another in future.
Organisations must become comfortable with the fact that data migration is almost inevitable when managing digital records for +25 years. In the case of clinical data, that migration is going to be easiest close to the point of submission given the recent access to the locked system, and staff are familiar with those systems and the datasets involved. The further away from that point, the harder the migration will be, and the greater risk that issues may arise from the transfer.
4- Distinct active, locked and archival systems
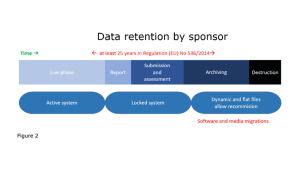
This diagram illustrates how data should be stored at each stage of the clinical trial. While the trial is live, data should be managed in an active system. After it has closed, it must be secured in a locked system for reporting, submission and assessment. Then finally for the archiving stage, data should be retained in such a state that the trial can be rebuilt if required.
It is important to reiterate that the EMA are depicting the lifecycle of trial data in three separate and distinct stages; active, locked and during archival.
They have also made it clear that ‘software and media migrations’ are likely (or even expected) – for data to endure for that length of time, data migration is also almost inevitable.
This is an indication that active and locked systems are not fit for purpose to archive data, and do not keep long-term records in line with the ALCOA++ principles. Arkivum would highly recommend migrating your clinical records into a dedicated archive soon after submission, to guarantee long-term access, use and most importantly, compliance.
5- “An inventory of all essential data and documents and corresponding retention periods should be maintained.”
This section is very straightforward and to the point. A list (inventory) must be kept of all documents and data collected with their corresponding retention times. Each retention period should be based on the applicable regulations surrounding it.
For instance, where the data is captured by electronic medical devices, the retention periods of this data must reflect EU medical device regulation requirements (EU Regulation 2017/745 to be precise).
6- “Retention periods should respect the data protection principle of storage limitation.”
Essentially, clinical trial data must be archived in line with data protection requirements. Personal data must not be kept for longer than is needed for its collection purpose, nor past the retention requirements. This is a key aspect of data protections laws (e.g. EU General Data Protection Regulation) which eliminates the misuse of personal data including retention for unnecessary lengths of time.
In following this guidance, organisations should specify in patient contracts how long their data will be held for (reflecting the archiving requirements) and then after the specified period, data must be erased (unless a regulatory board has given instruction not to).
In Summary…
The EMA guidelines paint a clear picture of how to comply with retention rules set out in regulations such as EU Reg 536/2014; alignment to the ALCOA++ principles throughout the entire data management lifecycle are crucial.
Other highlights from the guidelines include:
- The lifecycle of clinical data has three separate and distinct phases – live, locked and archived
- The migration of digital clinical records from a locked system are almost inevitable over the 25+ year retention period
- A suitable archive must be sought for long-term records
- And this archive must ensure the long-term access to those records.
Given the length of the guidance document, we’ve decided to dedicate a second post to some of the details held within the Annexes…we’ll therefore be publishing part 2 of this post soon.
Finally, if there is anything in the post you’d like to discuss with us further, please do not hesitate to get in touch here.

Caitlin Morris
Caitlin is the Content Marketing Manager at Arkivum. She joined the company in 2022 and is responsible for internal and external content creation and management. Caitlin has over 4 years of business and marketing experience.
Get in touch
Interested in finding out more? Click the link below to arrange a time with one of our experienced team members.
Book a demo